Addressing Clean Energy Challenges



Quantum Chemistry: Tackling complex problems beyond current computational limits.
AI for Generation: AI enhances material design for better energy storage.



GPU Utilization: Leveraging GPUs today through QEMIST Cloud for effective quantum and AI powered simulations.
QPU Readiness: Preparing for quantum computing with future integration of QPUs in Tangelo.

AI Limitations: AI is crucial but not sufficient alone for ultimate solutions.
Quantum Necessity: Quantum simulation is essential for accurate, foundational data.

Discover our solution where AI and Quantum advancements enhance lithium-based batteries, significantly improving energy efficiency and capacity across applications.
Incremental Full Configuration Interaction (iFCI) can offer quantitative predictions of molecular properties with high accuracy, which is essential for understanding the behavior of materials in battery systems.
For instance, iFCI calculations can accurately predict the electronic structure of molecules, aiding in the design of high-performance electrode materials.
Density Functional Theory (DFT) can efficiently calculate the electronic structure of molecules, including their energy levels, charge distributions, and bonding characteristics.
For materials useful as battery electrolytes, DFT can model solvent-solute interactions, helping to understand the solvation behavior and the impact of different solvents on battery performance.
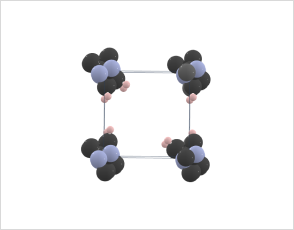
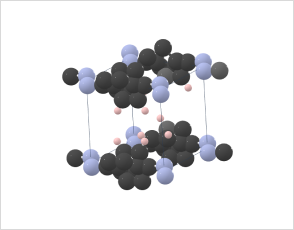
Molecules can exist in different crystal polymorphs, each with distinct packing arrangements of molecules in the crystal lattice.
Crystal structure prediction methods can explore the large set of possibilities and predict the most stable crystal structures under different conditions.
Need help choosing the right solver?
Discover the three solver types below:
In this demo, we will study a few examples of liquid and solid electrolytes.
The stability of electrolytes is important for battery capability, and the stability can be estimated based on the energy calculation from quantum chemistry.
For the liquid electrolytes (ethylene carbonate (EC) and propylene carbonate (PC)), we can use the Incremental Full Configuration Interaction (iFCI) and Density Functional Theory (DFT) solvers to estimate their stability.
When a high-accuracy estimation is required, we can use iFCI. Although its computational cost is high, iFCI gives us the most accurate energy calculation. …
Need help choosing the right solver?
Discover the three solver types below:
In this demo, we will study a few examples of liquid and solid electrolytes.
For the liquid electrolytes (ethylene carbonate (EC) and propylene carbonate (PC)), we can use the Incremental Full Configuration Interaction (iFCI) and Density Functional Theory (DFT) solvers to estimate their stability.
When a high-accuracy estimation is required, we can use iFCI. Although its computational cost is high, iFCI gives us the most accurate energy calculation. …
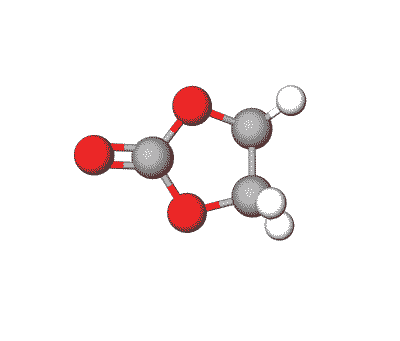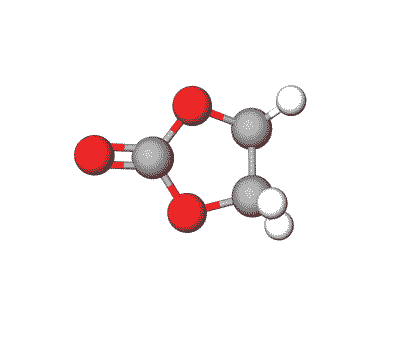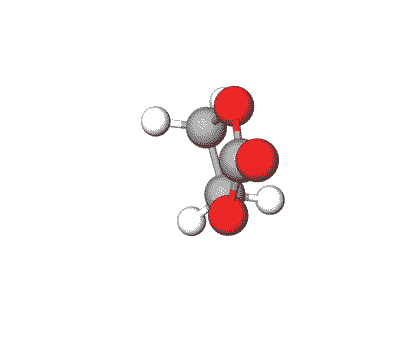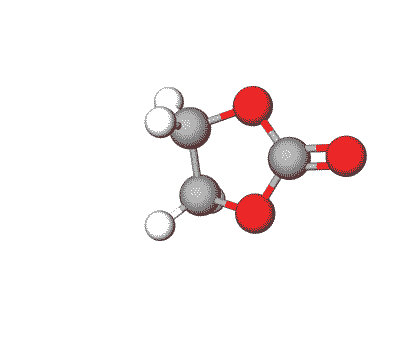
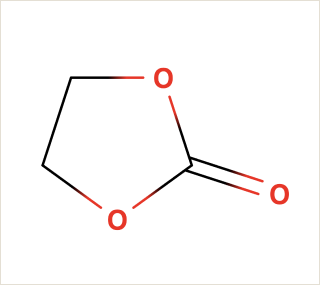
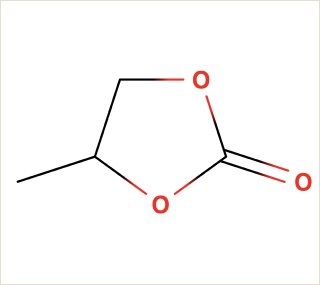
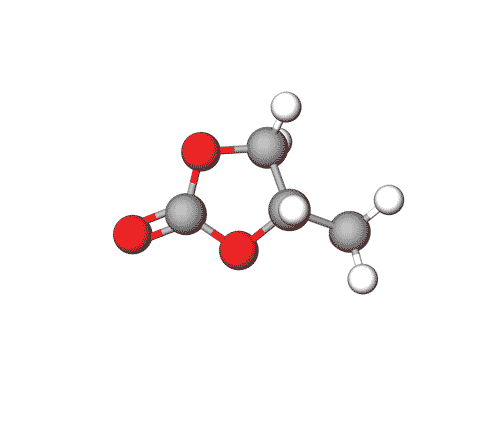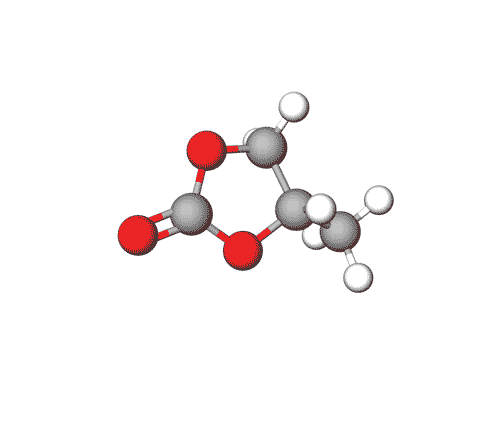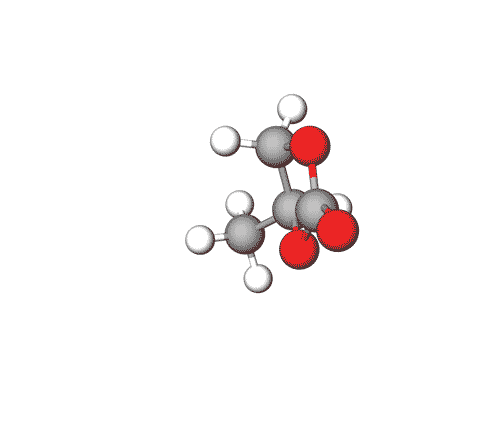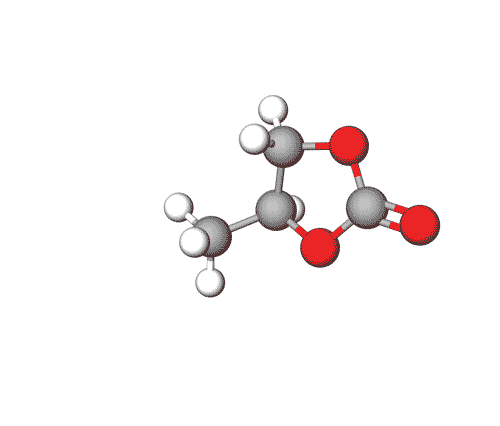
Ethylene Carbonate is commonly used as a polar solvent in various chemical processes, including in lithium-ion batteries as an electrolyte additive.
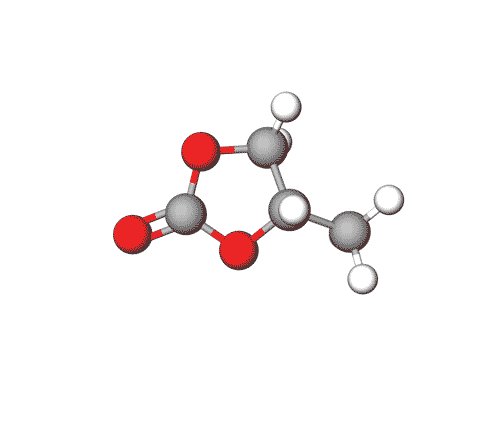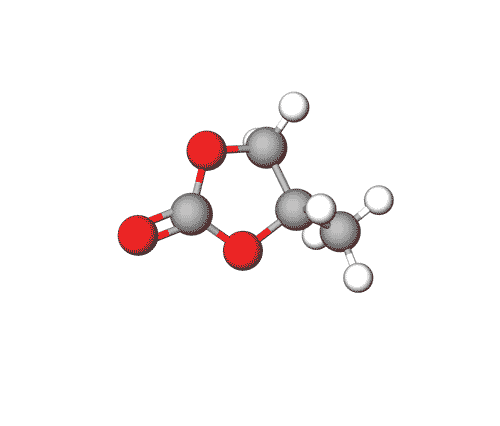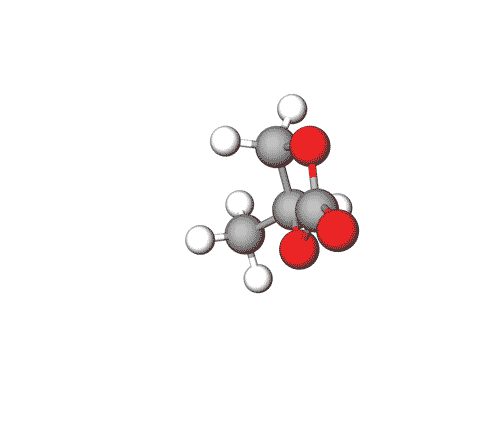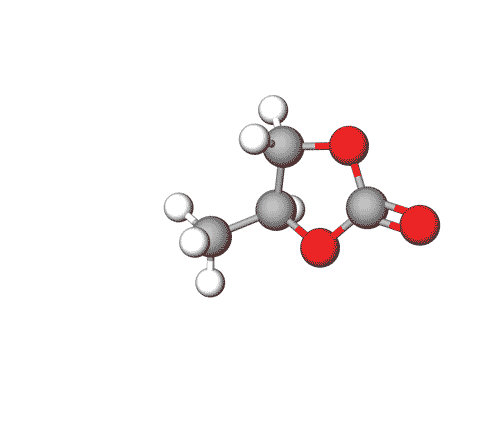
Propylene Carbonate can be used in electrolytes for lithium batteries, as a solvent for cellulose derivatives, and in other industrial applications.

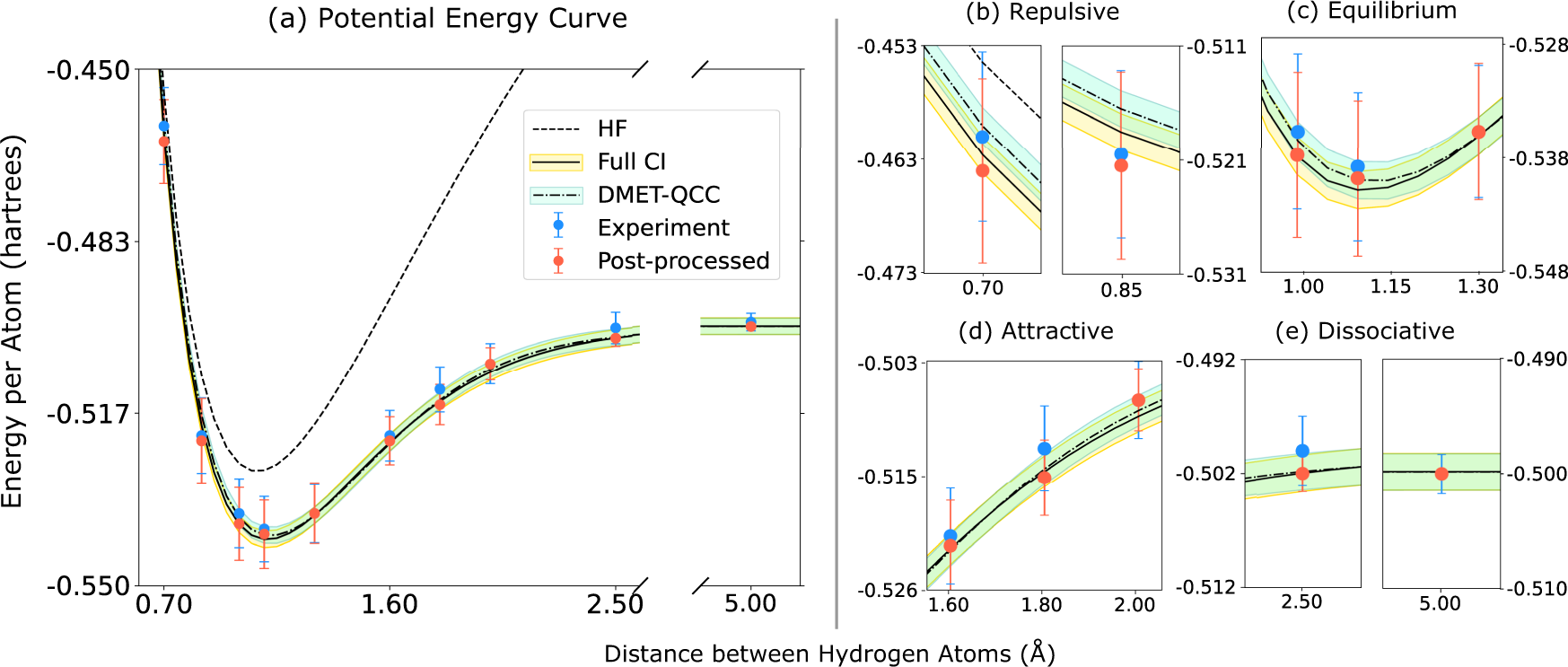
DOI: 10.1038/s42005-021-00751-9
Users will have the ability to study chemical systems using both classical and quantum computing, similar to choosing solvers in the first step of this demo. There are systems where classical computing is the appropriate choice, and there are systems that will be accessible thanks to evolving quantum computing methods.
In this plot, a hybrid quantum algorithm – qubit coupled-cluster ansatz and variational quantum eigensolver (DMET-QCC) – was run on a trapped-ion quantum computer to study a chemical system. DMET-QCC was shown to approach chemical accuracy and is an early demonstration of the potential for problem decomposition to accurately simulate large
| O | -0.0181 | 1.1195 | -0.1121 |
| O | -0.0183 | -1.1196 | 0.1115 |
| O | 1.9822 | -0.0001 | 0.0004 |
| C | -1.3529 | 0.7276 | 0.1723 |
| C | -1.3532 | -0.7274 | -0.1718 |
| C | 0.7603 | -0.0001 | -0.0003 |
| H | -2.0651 | 1.3016 | -0.4263 |
| H | -1.5483 | 0.9066 | 1.2348 |
| H | -2.0651 | -1.3012 | 0.4274 |
| H | -1.5495 | -0.9063 | -1.2342 |
When the field evolves to the point that quantum computing provides an advantage, researchers will be able to seamlessly run calculations on their choice of quantum computing hardware to study systems of interest.
DOI: 10.1038/s42005-021-00751-9
Users will have the ability to study chemical systems using both classical and quantum computing, similar to choosing solvers in the first step of this demo. There are systems where classical computing is the appropriate choice, and there are systems that will be accessible thanks to evolving quantum computing methods.
In this plot, a hybrid quantum algorithm – qubit coupled-cluster ansatz and variational quantum eigensolver (DMET-QCC) – was run on a trapped-ion quantum computer to study a chemical system. DMET-QCC was shown to approach chemical accuracy and is an early demonstration of the potential for problem decomposition to accurately simulate large
| O | -0.0218 | 0.9508 | 0.0405 |
| O | 0.7840 | -1.1505 | 0.0333 |
| O | 2.2453 | 0.6154 | -0.0951 |
| C | -0.0951 | 0.0865 | 0.3926 |
| C | -0.6257 | -1.2401 | -0.1262 |
| C | -0.1262 | 0.5592 | -0.2317 |
| C | 1.1072 | 0.1787 | -0.0134 |
| H | -1.1805 | 0.0841 | 1.4860 |
| H | -1.0253 | -2.0793 | 0.4501 |
| H | -0.8432 | -1.3980 | -1.1881 |
| H | -2.2992 | 0.6471 | -1.3198 |
| H | -3.2155 | -0.1244 | -0.0025 |
| H | -2.6511 | 1.5567 | 0.1397 |
When the field evolves to the point that quantum computing provides an advantage, researchers will be able to seamlessly run calculations on their choice of quantum computing hardware to study systems of interest.
Ethylene Carbonate is commonly used as a polar solvent in various chemical processes, including in lithium-ion batteries as an electrolyte additive.
Propylene Carbonate can be used in electrolytes for lithium batteries, as a solvent for cellulose derivatives, and in other industrial applications.
DOI: 10.1038/s42005-021-00751-9
Users will have the ability to study chemical systems using both classical and quantum computing, similar to choosing solvers in the first step of this demo. There are systems where classical computing is the appropriate choice, and there are systems that will be accessible thanks to evolving quantum computing methods.
In this plot, a hybrid quantum algorithm – qubit coupled-cluster ansatz and variational quantum eigensolver (DMET-QCC) – was run on a trapped-ion quantum computer to study a chemical system. DMET-QCC was shown to approach chemical accuracy and is an early demonstration of the potential for problem decomposition to accurately simulate large
| O | -0.0181 | 1.1195 | -0.1121 |
| O | -0.0183 | -1.1196 | 0.1115 |
| O | 1.9822 | -0.0001 | 0.0004 |
| C | -1.3529 | 0.7276 | 0.1723 |
| C | -1.3532 | -0.7274 | -0.1718 |
| C | 0.7603 | -0.0001 | -0.0003 |
| H | -2.0651 | 1.3016 | -0.4263 |
| H | -1.5483 | 0.9066 | 1.2348 |
| H | -2.0651 | -1.3012 | 0.4274 |
| H | -1.5495 | -0.9063 | -1.2342 |
When the field evolves to the point that quantum computing provides an advantage, researchers will be able to seamlessly run calculations on their choice of quantum computing hardware to study systems of interest.
Users will have the ability to study chemical systems using both classical and quantum computing, similar to choosing solvers in the first step of this demo. There are systems where classical computing is the appropriate choice, and there are systems that will be accessible thanks to evolving quantum computing methods.
In this plot, a hybrid quantum algorithm – qubit coupled-cluster ansatz and variational quantum eigensolver (DMET-QCC) – was run on a trapped-ion quantum computer to study a chemical system. DMET-QCC was shown to approach chemical accuracy and is an early demonstration of the potential for problem decomposition to accurately simulate large
| O | -0.0218 | 0.9508 | 0.0405 |
| O | 0.7840 | -1.1505 | 0.0333 |
| O | 2.2453 | 0.6154 | -0.0951 |
| C | -0.0951 | 0.0865 | 0.3926 |
| C | -0.6257 | -1.2401 | -0.1262 |
| C | -0.1262 | 0.5592 | -0.2317 |
| C | 1.1072 | 0.1787 | -0.0134 |
| H | -1.1805 | 0.0841 | 1.4860 |
| H | -1.0253 | -2.0793 | 0.4501 |
| H | -0.8432 | -1.3980 | -1.1881 |
| H | -2.2992 | 0.6471 | -1.3198 |
| H | -3.2155 | -0.1244 | -0.0025 |
| H | -2.6511 | 1.5567 | 0.1397 |
When the field evolves to the point that quantum computing provides an advantage, researchers will be able to seamlessly run calculations on their choice of quantum computing hardware to study systems of interest.
Azobenzene and its derivatives can exist in multiple crystal polymorphs, each with distinct packing arrangements of molecules in the crystal lattice.
Predicting polymorphs, CSP helps understand the crystal packing motifs and the factors influencing polymorphic transitions, which are crucial for applications such as materials design and understanding photoresponsive properties.
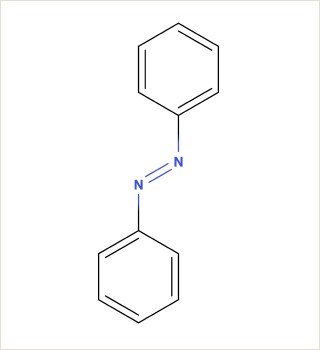
Customizing electrolyte formulas for better battery performance.
Speeding up development through streamlined experiments.
Creating innovative, patent-ready electrolyte blends.
Investigating a wide array of material combinations.
Filtering out less effective materials quickly.
Identifying the best candidates for further development.
Enhancing accuracy in battery life predictions.
Adapting models to improve battery longevity.
Reducing experimental costs and time.


